MESS (Massive Eco-Evolutionary Synthesis Simulations) - Part II
This is the second part of the MESS tutorial in which we introduce the API mode using jupyter notebooks. In this tutorial we’ll walk through the entire simulation and analysis process. This is meant as a broad introduction to familiarize users with the general workflow, and some of the parameters and terminology. We will use as an example in this tutorial the spider community data set from La Reunion published by Emerson et al (2017). However, you can follow along with one of the other example datasets if you like, the procedure will be identical although your results will vary.
MESS Part II Outline
- Crash course in machine learing
- Setting up and connecting to a notebook server on the cluster
- Create and parameterize a new MESS Region
- Run MESS simulations in API mode
- ML assembly model classification
- ML parameter estimation
- Posterior predictive simulations
- Free time to experiment with other example datasets
For the purposes of this tutorial, all command interactions will take place inside a jupyter notebook running on the cluster. For the most part we will be writing and executing python commands.
Crash course in machine learning
Here, Megan will introduce you to Random Forest, how it works, and what we can use it for. random forest presentation
The MESS.inference architecture is based on the powerful and extensive
scikit-learn python machine learning library. Another really amazing resource I highly recommend is the
Python Data Science Handbook.
Setting up and connecting to a notebook server on the cluster
Lets get set up and connected to our notebook server on the cluster again. If you get stuck you might review the jupyter notebook troubleshooting page,
Assuming your SSH tunnel is still running, you will only need open your web
browser and type localhost:<your_port_#>. Once you are connected to your
notebook server, navigate to ~/MESS` and choose “New->Notebook->Python 3”
Download and examine example data
We will be using as an example dataset of community-scale COI sequences (~500bp) and densely sampled abundances for the spider community on the island of La Reunion, an overseas department of France, which is the largest of the Mascarene islands, located in the Indian Ocean approximately 1000 km from Madagascar. The data we will be using was collected and published by Emerson et al (2017). For this exercise, we will just grab and use the formatted data from the MESS github repository. For further instruction on properly formatting and converting raw data into MESS-ready format see the MESS raw data handling page.
In a new cell in your notebook you can download the Reunion spider data like this:
!wget https://raw.githubusercontent.com/messDiv/MESS/master/empirical_data/Reunion_spiders/spider.dat
NB: The
!prior to the command here indicates that the jupyter notebook should interpret this as a bash command executed at the command line. This is a handy way of executing bash commands from within the notebook environment, rather than returning to a terminal window on the cluster. It’s just a nice shortcut.
Now make a new cell and import MESS and pandas (which is a python structured data library providing the DataFrame class), and read in the data you just downloaded.
%matplotlib inline
import MESS
import pandas as pd
spider_df = pd.read_csv("spider.dat", index_col=0)
spider_df[:5]
Special Note: The
%matplotlib inlinecommand tells jupyter to draw the figures actually inside your notebook environment. If you don’t put this your figures won’t show up!
NB: Importing pandas as
pdis pretty cannonical. It makes typing out pandas commands shorter because you can reference it aspdrather thanpandas.
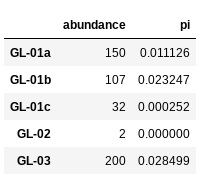
NB: The final line in the above command asks python to display the first 5 rows of the
spider_dfdataframe. It should look like this:
Create and parameterize a new MESS Region
MESS API mode lets you dive under the hood of the CLI mode a bit. You have
all the power of the CLI mode, yet more flexibility. The first step in API
mode is to create a MESS Region. A Region encompasses a very large
Metacommunity and a much smaller local community which is connected to it
by colonization. In creating a Region the only thing you’re required to
pass in is a name, so lets go with “LaReunion” (No spaces!), as this is the
region the empirical data is drawn from.
reunion = MESS.Region("LaReunion")
print(reunion.get_params())
------- MESS params file (v.0.1.0)----------------------------------------------
LaReunion ## [0] [simulation_name]: The name of this simulation scenario
./default_MESS ## [1] [project_dir]: Where to save files
0 ## [2] [generations]: Duration of simulations. Values/ranges Int for generations, or float [0-1] for lambda.
neutral ## [3] [community_assembly_model]: Model of Community Assembly: neutral, filtering, competition
point_mutation ## [4] [speciation_model]: Type of speciation process: none, point_mutation, protracted, random_fission
2.2e-08 ## [5] [mutation_rate]: Mutation rate scaled per base per generation
2000 ## [6] [alpha]: Abundance/Ne scaling factor
570 ## [7] [sequence_length]: Length in bases of the sequence to simulate
------- Metacommunity params: --------------------------------------------------
100 ## [0] [S_m]: Number of species in the regional pool
750000 ## [1] [J_m]: Total # of individuals in the regional pool
2 ## [2] [speciation_rate]: Speciation rate of metacommunity
0.7 ## [3] [death_proportion]: Proportion of speciation rate to be extinction rate
2 ## [4] [trait_rate_meta]: Trait evolution rate parameter for metacommunity
1 ## [5] [ecological_strength]: Strength of community assembly process on phenotypic change
------- LocalCommunity params: Loc1---------------------------------------------
Loc1 ## [0] [name]: Local community name
1000 ## [1] [J]: Number of individuals in the local community
0.01 ## [2] [m]: Migration rate into local community
0 ## [3] [speciation_prob]: Probability of speciation per timestep in local community
These are all the parameters of the model. The defaults are chosen to reflect
a typical oceanic island arthropod community. Don’t worry at this point about
all the parameters, lets focus for now on community_assembly_model, the
size of the local community (J), and the rate of migration from the metacommunity
to the local community (m). We will set parameter ranges for these, and each
simulation will sample a random value from this range. In a new cell use the
set_param() method to change these values:
reunion.set_param("community_assembly_model", "*")
reunion.set_param("J", "1000-10000")
reunion.set_param("m", "0.001-0.01")
NB: Setting the
community_assembly_modelto*indicates that we want to sample uniformly among all three of the model types: neutral, competition, and environmental filtering.
Print the params again to prove to yourself that the ranges are now set:
print(reunion.get_params())
------- MESS params file (v.0.1.0)----------------------------------------------
LaReunion ## [0] [simulation_name]: The name of this simulation scenario
./default_MESS ## [1] [project_dir]: Where to save files
0 ## [2] [generations]: Duration of simulations. Values/ranges Int for generations, or float [0-1] for lambda.
* ## [3] [community_assembly_model]: Model of Community Assembly: neutral, filtering, competition
point_mutation ## [4] [speciation_model]: Type of speciation process: none, point_mutation, protracted, random_fission
2.2e-08 ## [5] [mutation_rate]: Mutation rate scaled per base per generation
2000 ## [6] [alpha]: Abundance/Ne scaling factor
570 ## [7] [sequence_length]: Length in bases of the sequence to simulate
------- Metacommunity params: --------------------------------------------------
100 ## [0] [S_m]: Number of species in the regional pool
750000 ## [1] [J_m]: Total # of individuals in the regional pool
2 ## [2] [speciation_rate]: Speciation rate of metacommunity
0.7 ## [3] [death_proportion]: Proportion of speciation rate to be extinction rate
2 ## [4] [trait_rate_meta]: Trait evolution rate parameter for metacommunity
1 ## [5] [ecological_strength]: Strength of community assembly process on phenotypic change
------- LocalCommunity params: Loc1---------------------------------------------
Loc1 ## [0] [name]: Local community name
1000-10000 ## [1] [J]: Number of individuals in the local community
0.001-0.01 ## [2] [m]: Migration rate into local community
0 ## [3] [speciation_prob]: Probability of speciation per timestep in local community
Run MESS simulations in API mode
Now we can run a bunch of simulations given our new parameterization using the
run() method. Run takes one argument (nsims) which indicates the number
of independent community assembly realizations to perform.
reunion.run(sims=1)
Generating 1 simulation(s).
[####################] 100% Finished 0 simulations | 0:00:00 |
We will not do this now, but the
runmethod can also accept anipyclientargument for specifying a connection to an ipyparallel backend, allowing for massive parallelization. For more info see the MESS parallelization documentation.
Since it can take quite some time to run a number of simulations sufficient for
model selection and parameter estimation we will use a suite of pre-baked
simulations I generated ahead of time. Fetch them with wget from the compphylo
site:
!wget https://compphylo.github.io/Oslo2019/MESS_files/MESS_simulations/SIMOUT.txt
!wc -l SIMOUT.txt
100%[======================================>] 14,234,338 50.0M/s in 0.3s
2019-08-11 01:25:27 (50.0 MB/s) - "SIMOUT.txt.1" saved [14234338/14234338]
24440 SIMOUT.txt
NB: The
wccommand counts the number of lines if you pass it the-lflag. You can see this series of ~25k simulations is about 14MB.
ML assembly model classification
The first step is now to assess the model of community assembly that best
fits the data. The three models are neutral, in which all individuals are
ecologically equivalent; competition, in which species have traits, and
differential survival probability is weighted by distance of traits from
the trait mean in the local community (closer to the trait mean == higher
probability of death); and filtering, in which there is an environmental
optimum, and proximity of trait values to this optimum is positively
correlated with survival probability.
Basically we want to know, are individuals in the local community ecologically equivalent, and if not are they interacting more with each other or more with the local environment.
Here we create a MESS.inference.Classifier object and pass in the empirical
dataframe, the simulations, and specify RandomForest (‘rf’) as the algorithm
to use (other options are GradientBoosting (‘gb’) and AdaBoost (‘ab’)). Very
thorough documentation of all the machine learning inference architecture is
available on the MESS documentation site.
cla = MESS.inference.Classifier(empirical_df=spider_df, simfile="SIMOUT.txt", algorithm="rf")
est, proba = cla.predict(select_features=False, param_search=False, quick=True, verbose=True)
The Classifier.predict() method provides a very sophisticated
suite of routines for automating machine learning performance tuning.
Here we disable all of this in order for it to run quickly, trading
performance for accuracy. The select_features argument controls whether
or not to prune summary statistics based on correlation and informativeness.
The param_search argument controls whether or not to exhaustively search
the space of parameters to maximize accuracy of the ML algorithm selected.
Finally, the quick argument subsets the size of the simulation database
to 1000 simulations. In normal conditions, when you want to investigate
real data you should use select_featers=True, param_search=True, quick=False.
This will result in much more accurate inference, yet will take much more
time to run.
The call to cla.predict() returns both the most likely community assembly
model (est) and the model probabilities (proba), which you can view:
from IPython.display import display
display(est, proba)
NB: The first line of this codeblock imports the
displaymethod from theIPythonlibrary, which just makes pd.DataFrames pretty print.

Here we see that the neutral model is vastly favored over the competition
and fitering models.
Now we can examine the weight of importance of each feature, an indication
of the contribution of each summary statistic to the inference of the ML
algorithm. You can also get a quick and dirty plot of feature importances
directly from the Classifier object with the plot_feature_importances()
method.
display(cla.feature_importances())
cla.plot_feature_importance(figsize=(5,5))
NB: The
figsizeargument controls the size of the output figure. The default is(10, 10), so here we just make it a bit smaller to fit better in the nb window.
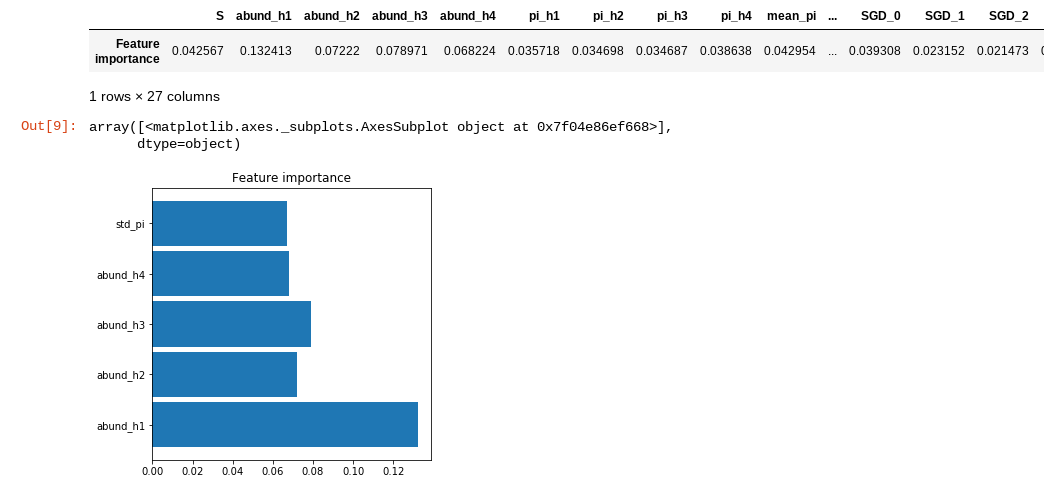
The plot_feature_importance() method prunes out all the summary statistics
that contribute less than 5% of information to the final model. This method is
simply for visualization. Here you can see that the shape of the abundance
distributions (as quantified by the first 4 hill numbers) and the standard
deviation of nucleotide diversity (pi) within the local community are the most
important summary statistics for differentiating between the different community
assembly models.
ML parameter estimation
Now that we have identified the neutral model as the most probable, we can estimate parameters of the emipirical data given this model. Essentially, we are asking the question “What are the parameters of the model that generate summary statistics most similar to those of the empirical data?”
The MESS.inference module provides the Regressor class, which has a very
similar API to the Classifier. We create a Regressor and pass in the
empirical data, the simulations, and the machine learning algorithm to use,
but this time we also add the target_model which prunes the simulations to
include only those of the community assembly model of interest. Again, we
call predict() on the regressor and pass in all the arguments to make it
run fast, but do a bad job.
rgr = MESS.inference.Regressor(empirical_df=spider_df, simfile="SIMOUT.txt", target_model="neutral", algorithm="rfq")
est = rgr.predict(select_features=False, param_search=False, quick=True, verbose=False)
NB: Note that here we ask for the
rfqalgorithm, which designates random forest quantile regression, and allows for constructing prediction intervals, something the stockrfalgorithm doesn’t do. The gradient boosting method (gb) provides prediction intervals natively.
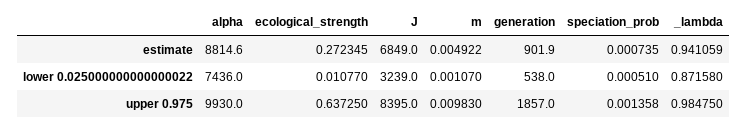
The return value est is a dataframe that contains predicted values for
each model parameter and 95% prediction intervals, which are conceptually
in the same ball park as confidence intervals and HPD.
display(est)
Similar to the classifier, you can also extract feature importances from the regressor, to get an idea about how each feature is contributing to parameter estimation. In this case, the feature importance plots are a little more useful than the giant table. Also note that we’re requesting a slighly larger figure, because this time there will be much more information.
display(rgr.feature_importances())
rgr.plot_feature_importance(figsize=(20,8))
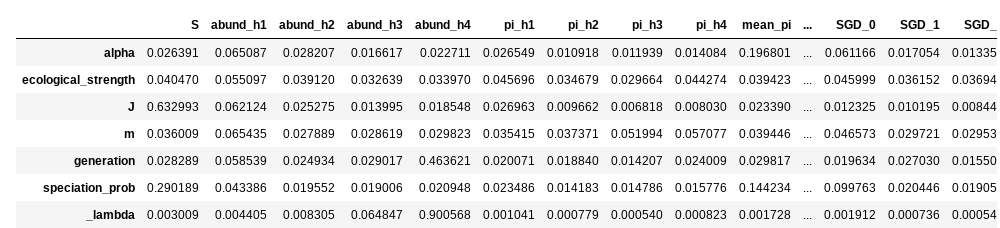
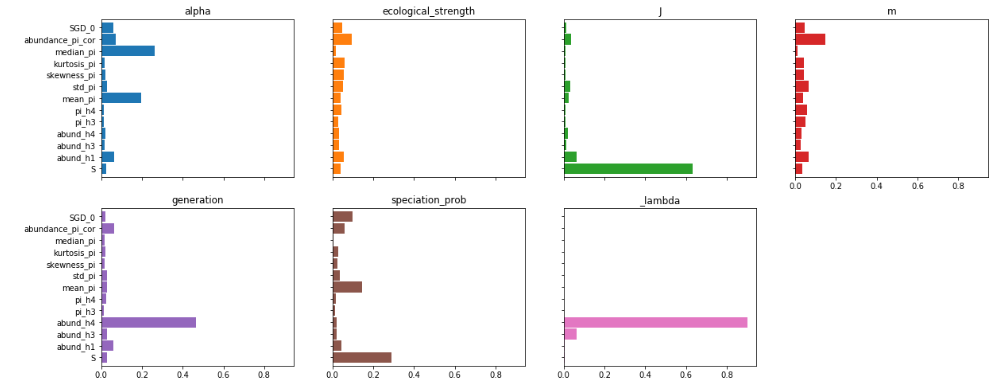
You can see that some parameters are strongly driven by one or a couple of
features, and for some the feature importances are more diffuse. For example
species richness (S) contributes overwhelmingly to estimation of local
community size (J), which makes sense. On the other hand, many more
factors seem to contribute equally to estimation of migration rate (m).
Perform posterior predictive simulations
Finally, a very important step in machine learning inference is to validate the parameter estimates by performing posterior predictive checks (PPCs). The logic of this procedure is that we will generate a suite of simulations using our most probable parameter estimates, and compare these simulations to our empirical data. For our purposes here we will project the summary statistics of observed and simulated datasets into principle component space. If the parameter estimates are a good fit to the data then our posterior predictive simulations will cluster together with the real data, whereas if the parameter estimates are a poor fit, then the real data will be quite different from the simulations.
Spoiler alert: Posterior predictive simulations can take quite a while to run. With the parameters we get even with this toy data a reasonable number of sims could take a couple hours. So! For the purpose of this exercise we are going to munge the estimates to make the simulations run faster. DO NOT DO THIS WITH YOUR REAL DATA!
est["J"] /= 2
est["m"] *= 10
est["_lambda"] /= 2
est
Here we reduced the size of the local community (J), increased the rate of
migration (m), and decreased the duration of the simulation (_lambda).
All these things will make the simulations run more quickly. Now go ahead
and run this next cell, it should take one to two minutes.
MESS.inference.posterior_predictive_check(empirical_df=spider_df,
parameter_estimates=est,
est_only=True,
nsims=20,
verbose=True,
force=True)
[# ] 5% Performing simulations
NB: The
posterior_predictive_check()function can also accept anipyclientparameter to specify to use an ipyparallel backend. For simplicity here we will not use this option, but in reality it would be a good idea to parallelize the posterior predictive simulations. See the MESS parallelization docs for more info about how to implement the parallel backend.
This time the only thing we have to pass in is the empirical data and
the dataframe of prediction values, but I’m showing a couple more arguments
here for the sake of completeness. Setting est_only to True will use only
the exact parameter estimates for all simulations, whereas setting it to
False will sample uniformly between the upper and lower prediction interval
for each simulation. nsims should be obvious, the number of posterior
predictive simulations to perform. force will overwrite any pre-existing
simulations, thus preventing mixing apples with oranges from different
simulation runs. On the other hand if this is False (the default), then
this is a handy way to add more PPCs to your analysis.
Here is a pretty typical example of a good fit of parameters to the data:
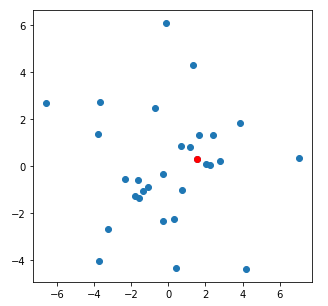
And here is a nice example of a poor fit of parameters to the data:
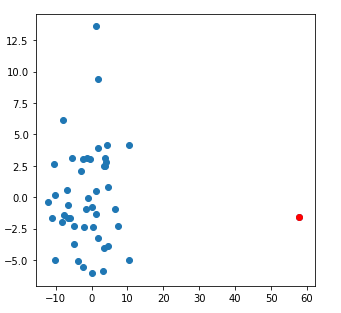
Quite a striking, and obvious difference.
Free time to experiment with other example datasets
Assuming there is any time left you might want to experiment either with some of the other empirical datasets which are provided on the MESS github repo, or with exploring the substantial MESS inference procedure documentation where we show a ton of other cool stuff MESS is capable of that we just don’t have time to go over in this short workshop.
References
Emerson, B. C., Casquet, J., López, H., Cardoso, P., Borges, P. A., Mollaret, N., … & Thébaud, C. (2017). A combined field survey and molecular identification protocol for comparing forest arthropod biodiversity across spatial scales. Molecular ecology resources, 17(4), 694-707.